
Dr. Ashley Kiemen, PhD / 2023 Research
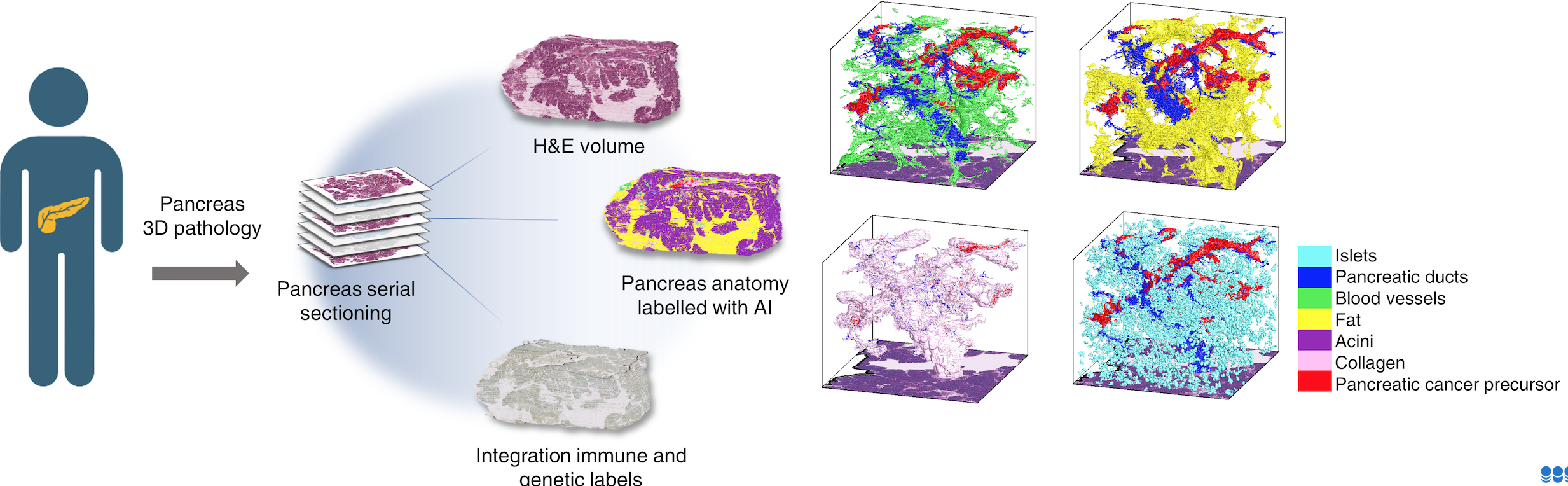
Dr. Ashley Kiemen, Ph.D. is an assistant professor of pathology at the Johns Hopkins University School of Medicine. She holds a master’s degree in philosophy and a doctorate in chemical & biomolecular engineering. Dr. Kiemen’s research focuses on development of computational tools for study of pancreatic cancer in three dimensions. Her projects have increased our understanding of the fundamental biology underlying the behavior of pancreatic cancer cells in the body. Specifically, she seeks to understand the structure, genetics, and immune microenvironment of pancreatic cancer precursor lesions, and to understand the means by which pancreatic cancer cells hijack existing structures in the pancreas to invade far from the primary tumor. Specifically, her team develops novel artificial intelligence-based tools to create digital maps of surgically resected pancreatic tumors, allowing researchers unparalleled views into the microanatomy of tumors. She hopes that the biology inferred from these maps will aid in development of screening tests that will allow us to detect pancreatic cancers earlier.
James R. Eshleman Jr., M.D. Ph.D Professor of Pathology and Oncology at Johns Hopkins
- Chemotherapy of pancreatic cancer
The Eshelman team recently developed a 3-dimensional model of pancreatic cancer and screened a library of 2700 drugs, mostly FDA approved for other diseases
- Molecular Relapse (minimal residual disease) test development in a clinical laboratory
The goal of this research is to validate a clinical test that detects circulating tumor DNA (ctDNA) in the plasma of pancreatic cancer patients and to validate ctDNA as a viable means of monitoring pancreatic cancer patients who undergo surgical resections to predict recurrence of disease. The team’s expectation is that, following surgery, the levels of ctDNA in the plasma fall to zero. The team hopes to show that persistant or rising levels of ctDNA in patients will predict future recurrence, allowing oncologists to know when a particular therapy is no longer working (due to rising levels) so a more effective therapy can be implemented.
Progress Report
Characterization of the Moment of Invasion in Pancreatic Neoplasms
Department of Pathology, Johns Hopkins University School of Medicine
Invasive pancreatic cancer is an aggressive deadly cancer with a dismal prognosis. However, invasive pancreatic cancer arises from non-invasive precursor lesions that, if detected early enough, are curable. Some of these precursor lesions are only visible microscopically, while others form large cysts that are detectable using currently available imaging technologies. While the goal is to remove these precancerous cysts before they progress to an invasive cancer, in some cases the resected precancerous cysts harbor a small associated invasive pancreatic cancer – these lesions provide a unique opportunity to understand the process of malignant transformation.
The goal of the work in our laboratory in the previous year has been to characterize the moment of transition between the non-invasive cyst and the infiltrating cancer, the moment in which a curable precursor lesion becomes an incurable invasive cancer. We are working to characterize this moment of invasion using two complementary cutting edge technologies – whole exome sequencing to identify genetic alterations and high throughput cell phenotyping (htCP) to identify phenotypic features.
In the past year, we have made significant progress towards the completion of our goal to characterize the moment of invasion in pancreatic cancer. We have developed a protocol to enrich for neoplastic tissue in paraffin blocks, followed by DNA extraction, library preparation, and next generation sequencing. We have performed a pilot study of 6 samples using this protocol and have obtained high quality whole exome sequencing data with an average 167 distinct reads per base and 93% of bases with >10X coverage. Based on mutation data, the average tumor cellularity was 63% in the sequenced samples. We identified an average of 64 mutated genes in the cancers and 43 mutated genes in the IPMNs – there was an average of 31 genes that were differentially mutated between the two components, including some genes with potential biological importance. We also have an additional 18 samples in our sequencing pipeline and expect data from these within the next several weeks.
In addition, in order to study tumor heterogeneity, we have developed a protocol for isolation and amplification of DNA from single pancreatic cancer cells. We have used this protocol to successfully perform molecular analyses of single cells from pancreatic cancer cell lines and are currently performing a rigorous analysis of the quality of the amplified DNA. Once we are confident that the amplified DNA is representative of that in a single cell, we will translate these techniques into individual patient samples.
We have also made significant progress in expanding high-throughput cell phenotyping (htCP) techniques to pancreatic neoplasms. We have applied this technique to formalin-fixed paraffin-embedded sections of pancreatic ductal adenocarcinoma, identifying several key phenotypic parameters that distinguish invasive carcinoma from non-neoplastic structures in the same sections. These preliminary studies have pinpointed crucial phenotypic parameters in pancreatic neoplasms and will provide a framework for interpretation of the phenotypic data in the current study of IPMNs.
Finally, we have developed a novel three-dimensional organoid culture model for pancreatic cancer and have successfully isolated and grown three-dimensional pancreatic cancer organoids from fresh human pancreatic cancer samples. These organoids invade when grown in collagen 1 gels, and using time lapse microscopy, the number of invasive foci can be quantified. This organoid culture model will be a critical tool in determining the functional effects of mutations identified in the whole exome sequencing of human tumor samples. It will allow us to distinguish mutations that are driving the invasive phenotype, as these mutations are the most promising targets for early detection and cancer prevention.
With data from these key preliminary genetic and phenotypic studies in hand, we will soon be able to translate our rigorously optimized protocols to analyze single cells from non-invasive IPMNs and associated minimally invasive carcinomas, providing key insights into the biology of early pancreatic cancers and identifying novel targets for the early detection of invasive pancreatic cancer.
Progress Report
Nicholas J. Robert Vet M.B. Ph.D.
December 2014
With the generous support of the Joseph C. Monastra Foundation we have made significant progress in the analysis of sequence data from the genomes of over 650 familial pancreatic cancers patients. These analyses have included:
- Identification of deleterious variants in known cancer susceptibility genes. We have identified all germline variants in over 70 genes that to predispose to cancer in our familial pancreatic cancer patients – these include known familial pancreatic cancer susceptibility genes and hereditary pancreatitis genes. Variants were then classified as deleterious, of unknown significance, or benign based on an in-house algorithm using population-based allele frequencies, functional predictions, and quality metrics. Burden of deleterious variants in the familial pancreatic cancer patients was compared to a set of controls to identify novel genes associated with familial pancreatic cancer.
- Filter-based analyses. We identified all deleterious variants (premature truncating and missense variants) in the coding regions of the familial pancreatic cancer patients. We then ranked genes based on the frequency of rare and private deleterious variants. Analysis of matched tumor to identify second somatic hits and family members to demonstrate segregation of deleterious variants with familial pancreatic cancer were used to narrow candidate lists. In this way, we identified and prioritized many candidate familial pancreatic cancer susceptibility genes.
- Tumor analyses. We have sequenced 34 tumors from whole-genome sequenced familial pancreatic cancer patients. Analysis of these tumors has identified somatic mutations and copy number alterations that occur during the development of pancreatic cancer. These data have been used to further prioritize candidate genes identified from filter-based analyses. Whole exome sequencing and analysis of 16 additional tumors is ongoing and will be integrated into analyses when complete.
Our successful analyses described above have identified many candidate genes for future in vitro and in vivo validation and characterization. These future studies will utilize organoids, a three-dimensional culture system derived from pancreatic ductal cells. First, we will establish cultures of pancreatic ductal organoids from normal and neoplastic ductal cells. Second, we will demonstrate efficient transduction of organoids using CRIPR/Cas9 gene-editing technology and validate their use as a familial pancreatic cancer model by modifying endogenous alleles of known familial pancreatic caner genes, for example BRCA2, PALB2, CDKN2A, TP53, and ATM. We will then characterize candidate susceptibility genes identified from bioinformatics analyses. These comprehensive analyses will identify the most important familial pancreatic cancer genes and have the potential to significantly impact patient care through personalized screening, early detection, and novel therapies.

